采用 ABACUS 进行表面计算(五):外加电场
作者:刘裕,邮箱:liuyu@stu.pku.edu.cn
审核:陈默涵,邮箱:mohanchen@pku.edu.cn
最后更新时间:2023/08/28
一、介绍
电场是电荷及变化磁场周围空间里存在的一种特殊物质。这种物质与通常的实物不同,它虽然不是由分子原子所构成的,但它却是客观存在的特殊物质,具有通常物质所具有的力和能量等客观属性。许多材料在外加电场作用下会表现出不同寻常的性质,例如:铁电体在外加电场的作用下会产生电极化,也可以通过施加的电场来控制磁性和自发极化,因此在信息储存和传输中有广泛应用;二维材料(如石墨烯)在外加电场下表现出不同的电子性质和可控制的物理性质,这种特性使其具有在电子学领域的广泛应用前景。在电子结构计算中,周期性边界条件下对体系施加电场较为容易,而施加磁场则困难很多。
因此,外加电场是密度泛函理论(Density Functional Theory,简称 DFT)软件中可以被实现且重要的功能。ABACUS 中实现了在表面体系中外加恒定电场的功能[1]。注意,在 ABACUS 中,外加电场功能存在相应的原子受力的修正,可以用于平面波(Plane-Wave)基组和数值原子轨道(Numerical Atomic Orbitals)基组计算,但注意该功能只能用于带真空层的表面体系。
本教程中将会展示如何在 ABACUS 计算中使用加电场功能,为使得这个教程较为有趣,我们举一个半金属二维材料 nanoribbon(见图 1)加电场产生半金属的例子来讲解如何给体系添加电场,该 DFT 的计算工作曾发在 2006 年的 Nature 杂志上,具体可参考文献[2]。在这个教程里,我们使用的是数值原子轨道基矢量(Numerical Atomic Orbitals),碳和氢的轨道半径都是 8 a.u.,但实际上也可以使用平面波基矢量,只是前者效率更高所以在这里采用。我们还会使用到电子的自洽场(self-consistent field,简称 SCF)计算,非自洽计算(non-self-consistent field,简称 NSCF),自旋非极化和极化计算等概念。在不考虑自旋时(即设置 nspin=1 时),计算出的能带图中没有带隙,为金属;考虑自旋之后,带隙打开,成为半导体;在自旋极化基础上,外加恒定电场之后,其中一个自旋表现为半导体,另一个自旋则为金属。
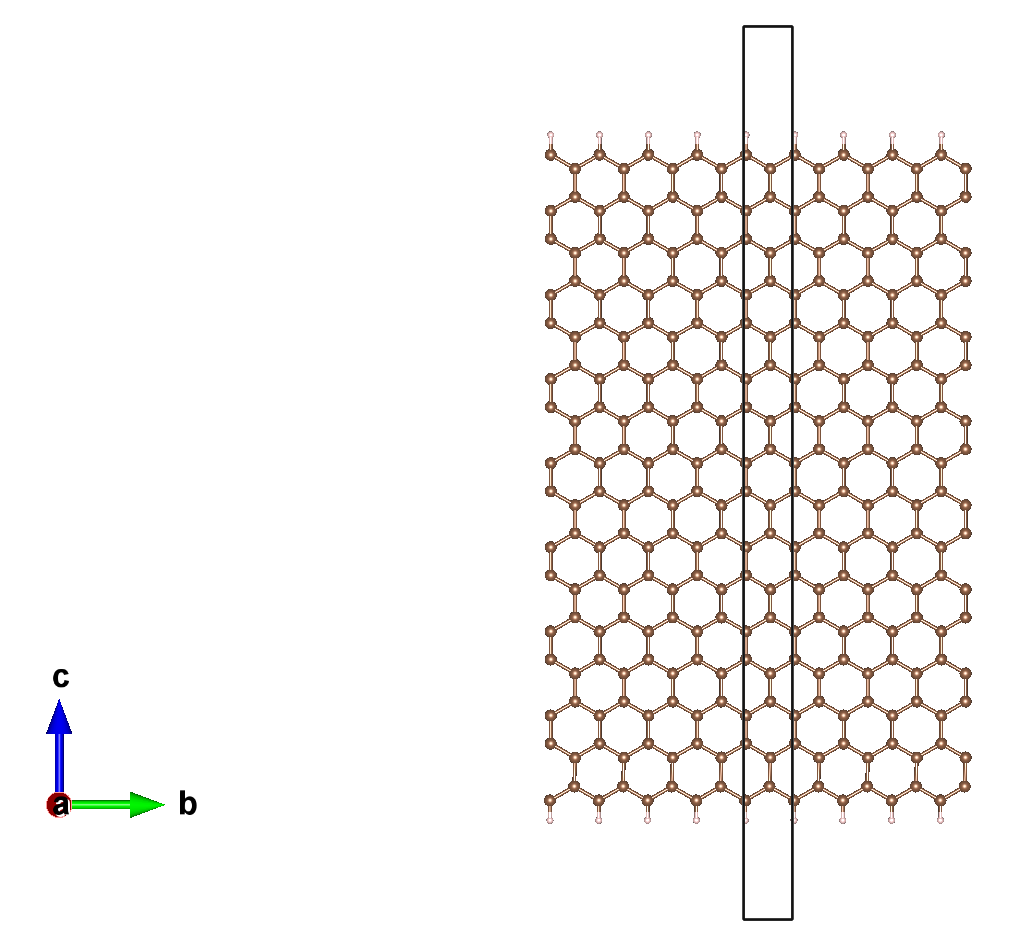
二、软件和算例准备
ABACUS 里关于外加电场的线上文档地址为:
http://abacus.deepmodeling.com/en/latest/advanced/scf/advanced.html#external-electric-field
ABACUS 里关于外加电场参数的线上文档地址为:https://abacus.deepmodeling.com/en/latest/advanced/input_files/input-main.html#electric-field-and-dipole-correction
ABACUS 的外加电场算例下载地址为(gitee):
https://gitee.com/MCresearch/abacus-user-guide/tree/master/examples/electric_field/
可以采用的下载命令是:
git clone https://gitee.com/MCresearch/abacus-user-guide
下载后解压,之后进入 /abacus-user-guide/examples/``electric_field 目录
或者采用 Github 仓库地址:
https://github.com/MCresearch/abacus-user-guide
三、采用加电场进行二维材料的能带计算
该算例中含有六个文件夹,解释如下:
1_unspin_scf:做非自旋极化(INPUT 文件中的 nspin 设为 1)的自洽场迭代 SCF 计算(INPUT 文件中的 calculation 设为 scf),输出自洽迭代之后的电子密度(将 INPUT 中的 out_chg 设为 1),注意二维材料的 KPT 文件,因为超胞大小是 102.459512146645 Å^3,所以对应的布里渊区 k 点取的是 1101,因为实空间周期长度小的方向对应的布里渊区大,所以需要取更多 k 点得到收敛结果。
2_unspin_band: 采用非自洽计算 NSCF(INPUT 文件中的 calculation 设为 nscf),读入上一个步骤输出的电荷密度(INPUT 文件中的 init_chg 设为 file),从而可以读入体系的基态电荷密度进行能带计算。
3_spin_scf:同 1_unspin_scf 的计算,只是把 nspin 设为 2。
4_spin_band:同 2_unspin_band 的计算,只是把 nspin 设为 2。
5_spin_elec_scf:同 3_spin_scf 的计算,只是又外加了电场的参数。
6_spin_elec_band:同 4_spin_band 的计算,只是又外加了电场的参数。
以下是算例 5 的输入文件(包含电场参数):
INPUT_PARAMETERS
#Parameters (General)
suffix nanoribbon
calculation scf
nbands 70
nspin 2
pseudo_dir ../../PP_ORB
orbital_dir ../../PP_ORB
#Parameters (Basis)
basis_type lcao
#Parameters (Accuracy)
ecutwfc 100
scf_thr 1e-6
scf_nmax 200
#Parameters (Smearing)
smearing_method gaussian
smearing_sigma 0.001
#Parameters (Charge mixing)
mixing_type pulay
mixing_beta 0.1
mixing_ndim 20
mixing_gg0 1.5
#Parameters (Efield)
efield_flag 1
efield_dir 2
efield_amp 0.0019440124
efield_pos_max 0.95
efield_pos_dec 0.10
以算例提供的 INPUT 文件为例,使用外加电场功能只需添加如下参数即可:
efield_flag:类型是 Bool,指定外加电场的开(1)或者关(0),默认值为 0efield_dir:类型是 Int,外加电场方向,可取 0,1,2 分别表示 x,y,z 方向,默认值为 2efield_pos_max:类型是 Real,锯齿状电势最大值所在分数坐标,范围[0,1),默认值为 0.5efield_pos_dec:类型是 Real,锯齿状电势从最大值到最小值的长度(同样以分数坐标为度量),默认值为 0.1,范围[0,1)。以图2为例,0.5 是最大值,0.6 是最小值,那么 0.5 ~ 0.6 的区域必须放置在没有原子的真空层中,注意这个例子设置的是 0.95 到 0.10 为电场区间。
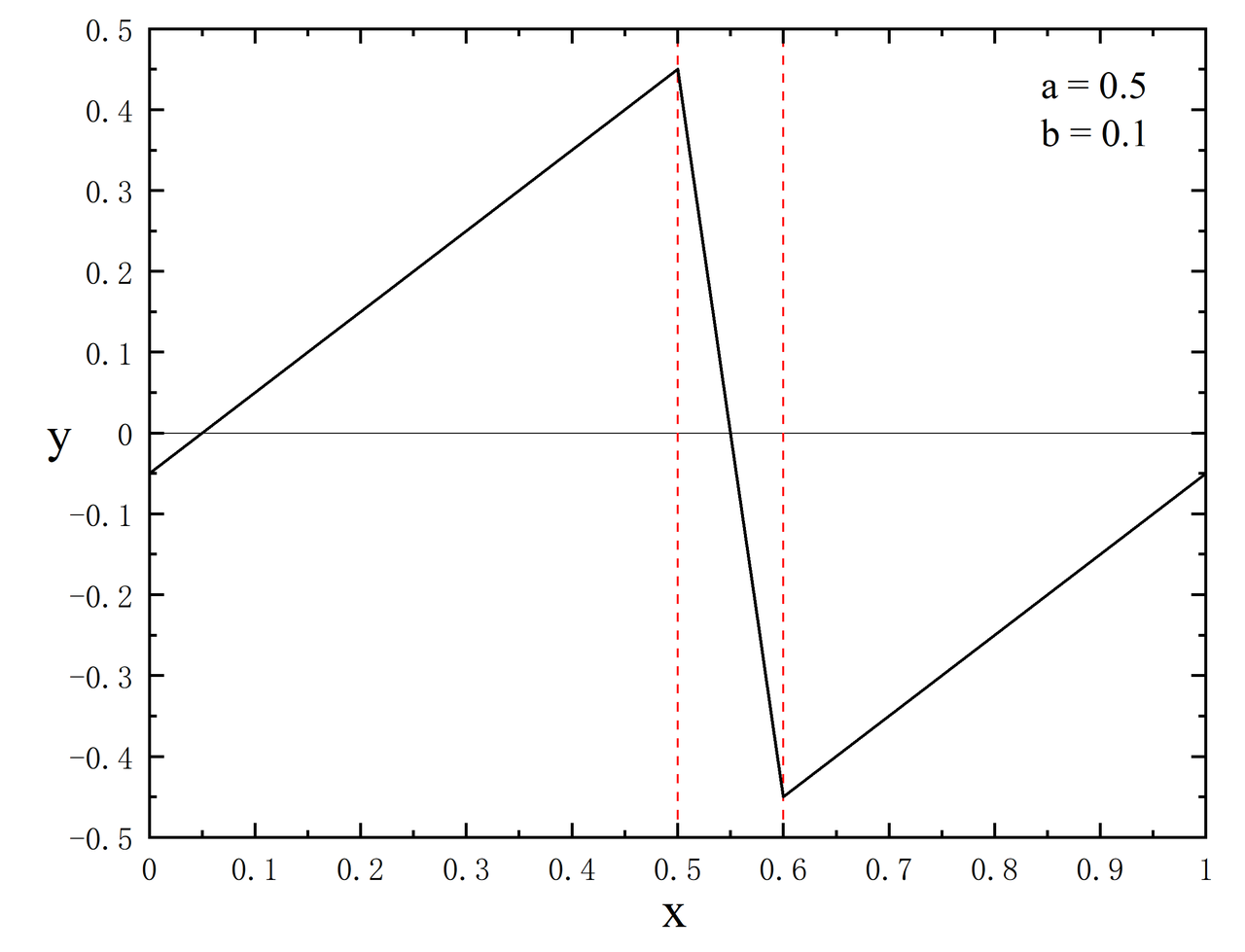
efield_amp:类型是 Real,电场强度,默认值为 0,单位为 a.u.,其中 1 a.u. = 51.4220632*10^10 V/m
以上参数的使用方法在采用 ABACUS 进行表面计算(二):偶极修正中有更详细的说明。
四、预期计算结果
- 运行完算例
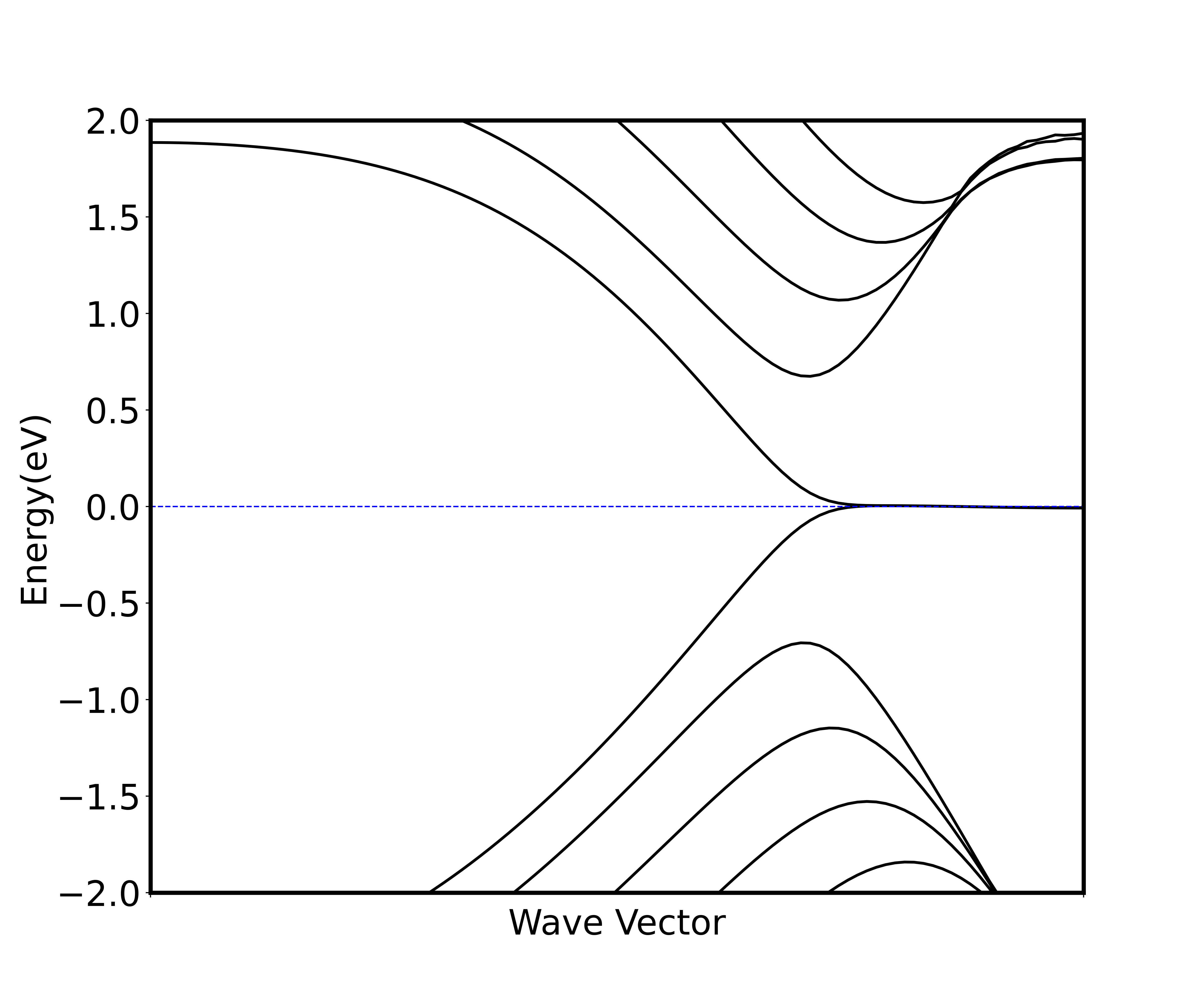
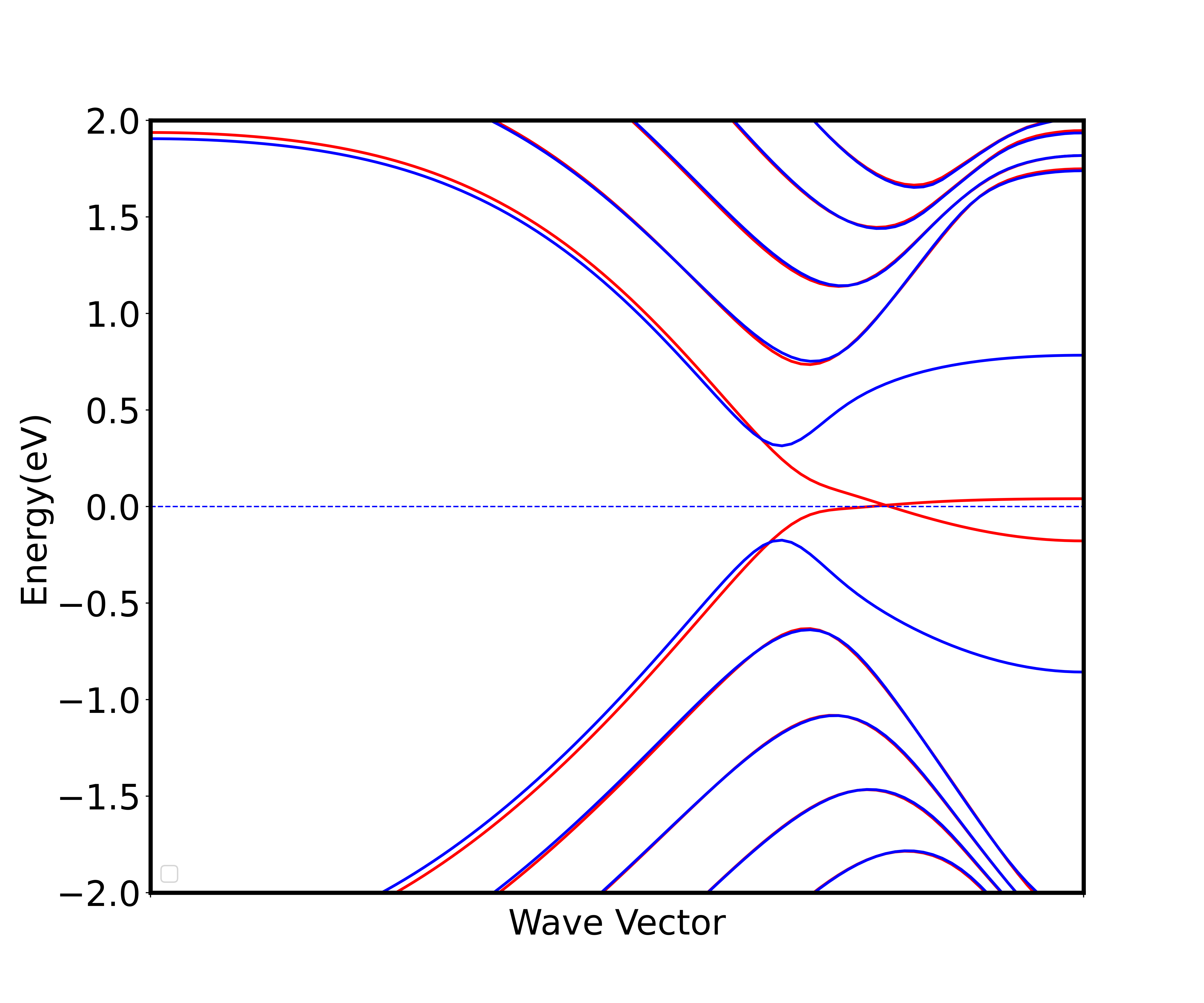
2_unspin_band后会通过非自洽计算得到体系非自旋极化能带图,横坐标是对二维材料布里渊区取k点,从Gamma点(坐标为0 0 0)取到点(坐标为0 0.5 0),取了 100 个k点算出来的能带图;纵坐标是能级,将费米面设置为 0,取了费米面附近 2 eV 的能量区间画出能带。以下两张图是同样取法。 - 运行完算例
4_spin_band后会得到非自洽计算得到的自旋极化能带图(图 3)。 - 运行完算例
6_spin_elec_band后会得到自旋极化加电场的能带图(图 4),注意这里所加的电场为 0.1 V/Å,由输入文件 INPUT 中的efield_amp设置,但efield_amp的单位是 a.u.,需要换算。
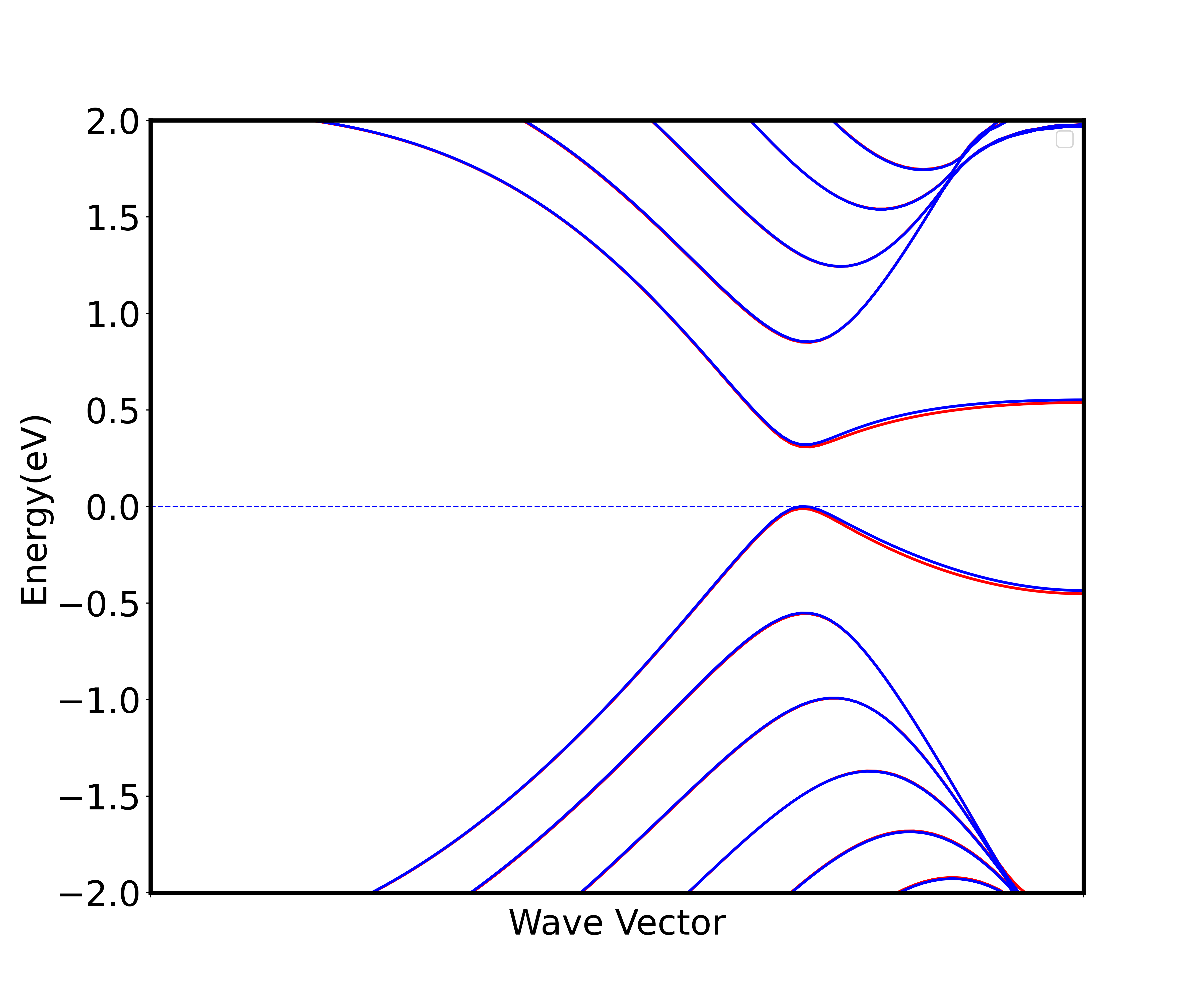
五、参考文献
[1] Phys. Rev. B 59, 12301 (1999), https://doi.org/10.1103/PhysRevB.59.12301
[2] Nature 444, 347–349 (2006), https://doi.org/10.1038/nature05180