ABACUS 实时演化含时密度泛函理论使用教程
作者:赵昊天,邮箱:zaotian@mail.ustc.edu.cn
审核:包涛尼,邮箱:baotaoni@pku.edu.cn
最后更新时间:2024 年 7 月 11 日
一、背景
实时演化含时密度泛函理论(Real-time Time-Dependent Density Functional Theory,简称 rt-TDDFT)是一种基于第一性原理的模拟激发态电子体系动态演化的方法。与传统的密度泛函理论(DFT)不同,rt-TDDFT 是建立在 Runge-Gross 定理之上,主要研究时间依赖的 Kohn-Sham 方程(TDKS 方程)。该理论通过构建时间传播子迭代求解 TDKS 方程,直接演化电子的波函数。此外,它还采用半经典的 Ehrenfest 动力学来模拟离子实的运动,可以实时地得到激发电子态以及离子位置的信息,因此具有较为广泛的应用前景。
周期性体系具有能带结构,而非周期性的原子或分子体系则具有离散能级。体系的基态是指价电子从低到高依次填充至能带或能级中。当体系吸收特定能量后,低能态的电子跃迁至高能态,此时体系便处于激发态。研究激发态性质是 TDDFT 的一大优势,因此掌握如何模拟体系的激发态变得尤为关键。ABACUS 为此提供了两种方式:一种是直接改变占据数,使得叠加态的波函数拥有更多高能态分量;另一种是向一个处于基态的体系施加一个外加电场,为其提供激发所需的能量。下面将分别介绍这两种方式的具体使用。文档中提到的算例可以在如下仓库中找到:https://gitee.com/mcresearch/abacus-user-guide/tree/master/examples/tddft
有关 TDDFT 输入参数的细节可参考输入参数线上文档:https://abacus.deepmodeling.com/en/latest/advanced/input_files/input-main.html#tddft-time-dependent-density-functional-theory
二、在ABACUS 中使用rt-TDDFT时改变电子的占据数
算例:https://gitee.com/mcresearch/abacus-user-guide/tree/master/examples/tddft/occupation
算例输入文件 INPUT 设置如下:
INPUT_PARAMETERS
#Parameters (1.General)
suffix H2_ocp
calculation md
esolver_type tddft
nbands 5
nspin 1
pseudo_dir ../../../tests/PP_ORB
orbital_dir ../../../tests/PP_ORB
#Parameters (2.Iteration)
ecutwfc 60
scf_thr 1e-6
scf_nmax 100
#Parameters (3.Basis)
basis_type lcao
gamma_only 0
#Parameters (4.Smearing)
smearing_method gauss
#Parameters (5.MD Parameters)
md_type nve
md_nstep 1000
md_dt 0.05
md_tfirst 0
#Parameters (6.TDDFT Occupation Parameters)
ocp 1
ocp_set 1 1 0 0 0
具体参数设置请参考线上文档,这里仅对部分重要参数进行说明:
gamma_only:必须设置为0,gamma_only为1会采用 double 类型计算,rt-TDDFT 使用传播子迭代计算波函数,需要 complex 类型存储数据,不兼容gamma_only为1的情况。basis_type:必须设置为lcao,目前 ABACUS 仅支持基于数值原子轨道基组的 rt-TDDFT 功能。nbands:能带数,由于ocp_set需要手动设置每一条能带的占据数到最高带,建议手动设置nbands,防止二者不对应。calculation:必须为md,同时一般 MD(Molecular Dynamics)计算所需的参数都需要设置,这部分参数会对应 rt-TDDFT 的离子实运动的计算。esolver_type:tddft,表示采用 rt-TDDFT 求解器。ocp:改变占据数功能的开关,为1的时候开启。ocp_set:占据数的设置,1 1 0 0`` 0表示这个算例所计算的五条能带,各自占据的电子数,其总和为 2,代表 H₂ 的两个价电子,可以简化为2*1 ``3``*0。另外,对于有多个 k 点的体系,ABACUS 支持对各个 k 点单独设置占据数。假设体系拥有 5 条能带,2 个 k 点,则ocp_set需要指定 10 个数值,前五个代表第一个 k 点五条能带的占据,后五个代表第二个 k 点五条能带的占据。
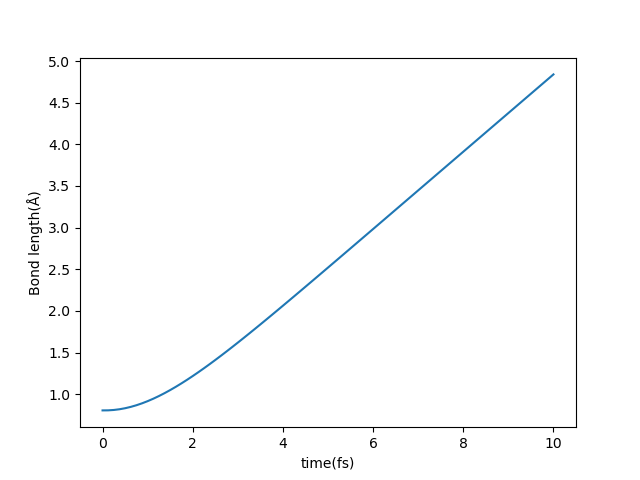
按照分子轨道理论,两个 H 原子的原子轨道发生相互作用形成一条成键轨道、一条反键轨道,H₂ 分子的基态占据可以表示为 2 0 0 0`` 0,最低的能态代表成键轨道,则 1 1 0 0`` 0 占据数表示将一个电子从成键轨道上激发到反键轨道,会导致 H₂ 分子发生解离。通过输出目录 OUT.H2_ocp/STRU 下的结构文件,可以看到随着时间的推移,两个 H 原子逐渐解离。运行 occupation 文件夹下的算例,通过 ASE-ABACUS 读取各个时刻 STRU 文件,并计算了两个 H 原子实时的间距,结果如下图所示:
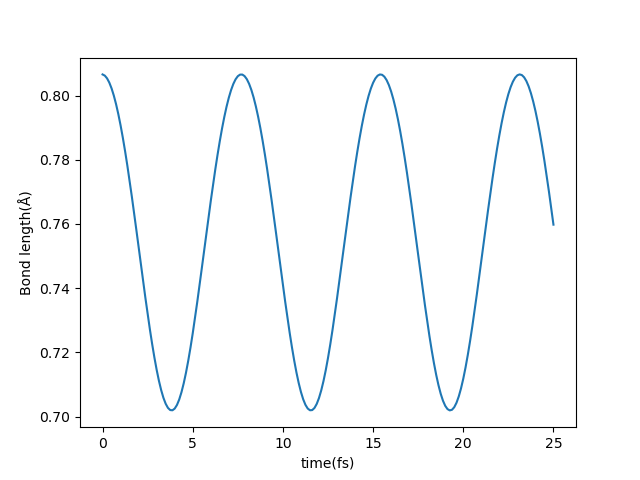
为了说明这个解离是由于改变占据数引起的,可以将 INPUT 替换为 occupation 文件夹中的 INPUT-1,其中 ocp_set 被改为:2 0 0 0`` 0,同样输出实时的原子间距,可以看到,两个原子间距稳定的振荡,并未发生解离:
三、ABACUS 中使用rt-TDDFT 外加电场计算材料吸收谱
在任意外加电场的影响下,rt-TDDFT 能够实时模拟并获得材料的电子态和离子位置信息。按照 Runge-Gross 定理(相当于 KSDFT 中的 Hohenberg-Kohn 定理),这等价于材料的全部物理性质。在众多的物理性质中,吸收谱作为材料基本的光电响应性质,处理简单,结果直观,可参考对比的数据丰富,很适合用于介绍 TDDFT 外加电场功能的基本细节。
3.1 规范选择
如何加入电场存在规范问题,根据电动力学的知识:
第一种是长度规范,把电场的影响归为纯标势的影响,向势场中加入:
因为晶胞很小,按照通常所用的光的波长,体系内的电场在空间上是近似匀强的,故这里没有写成积分的形式。这种形式相对简单,需要处理的项较少,程序处理较快,但是由于的存在,它在周期性体系的边界是不连续的,只能采用锯齿状的方式在边界下降来衔接这种不连续性。对于非周期性体系,边界存在真空层,这种锯齿状衔接不会影响到材料的响应,但是对周期性体系,这种做法就行不通了。
对于周期性体系必须采用速度规范,它用矢势而非标势来引入电场:
在这种规范下,需要对正常的项进行一系列变换,这里不详细论述,想进一步了解的可以参考文献 2 的内容。因而,我们实际上在程序里用的并不是直接的电场,而是标势或者矢势,但是它们都对应一个电场,程序指定电场的参数而不是势场的参数,这样可以统一处理长度和速度规范的场。
简单来说:长度规范只能处理非周期性体系,但是速度较快;速度规范可以处理周期性体系和非周期性体系,但是速度较慢,故一般建议对非周期性体系采用长度规范,周期性体系采用速度规范。
3.2 长度规范示例
算例:https://gitee.com/mcresearch/abacus-user-guide/tree/master/examples/tddft/absorption_H2_length
输入文件 INPUT:
INPUT_PARAMETERS
#Parameters (1.General)
suffix H2_absoprion
calculation md
esolver_type tddft
pseudo_dir ../../../tests/PP_ORB
orbital_dir ../../../tests/PP_ORB
#Parameters (2.Iteration)
ecutwfc 60
scf_thr 1e-6
scf_nmax 100
#Parameters (3.Basis)
basis_type lcao
gamma_only 0
#Parameters (4.Smearing)
smearing_method gauss
#Parameters (5.MD Parameters)
md_type nve
md_nstep 1000
md_dt 0.005
md_tfirst 0
#Parameters (6.Efield Parameters)
td_vext 1
td_stype 0
td_tstart 1
td_tend 1000
td_vext_dire 3 3
td_ttype 0 0
td_gauss_freq 3.66 1.22
td_gauss_phase 0.0 0.0
td_gauss_sigma 0.2 0.2
td_gauss_t0 300 300
td_gauss_amp 0.01 0.01
#Parameters (7.Output)
out_chg 1
out_efield 1
out_dipole 1
其他的注意事项和占据数一样,这里注意补充一些电场参数的细节:
md_nstep:MD 步,这里为了做演示只用了 1000 步,演化总时长 5 fs,为了精度考虑,实际计算时应当增加步数,具体视所需体系有所差异,一般保证 20 fs 以上总时长较为妥当。td_vext:外加电场的开关,1为打开外加电场。td_stype:规范的选择,0为长度规范,1为速度规范。td_tstart:开始加入电场的步数,在td_tstart步之前,为无电场状态,一般为1即可。td_tend:电场结束的步数,td_tend后不再计算电场,用于无电场时节约资源,最大可以等同md_nstep的值。td_tend后,标势为 0,而矢势固定为td_tend处的值不再变化。td_type:电场的波包形状,0-3分别对应高斯、梯形、三角、阶跃等不同类型的电场。td_gauss_xxx:由于这里指定的是高斯型,对应高斯型的参数,具体可以参考输入参数的说明文档:https://abacus.deepmodeling.com/en/latest/advanced/input_files/input-main.html#tddft-time-dependent-density-functional-theoryout_dipole:输出每个时刻的电偶极矩,我们通过对电偶极矩的分析得到吸收谱。out_efield:输出每个时刻的电场。
注意,这里为了覆盖更大的频率范围,通过对电场形式的参数多加一列的方式,在一次计算中加入了两个不同频率的电场。理论上可以用这种方式得到各种形状的场,当加的较多时,支持采用正则表达式 m*x 的形式进行简化。
运行算例,会输出全时刻的电偶极矩文件 SPIN1_DIPOLE,如果 nspin 为 2 则会有 SPIN2_DIPOLE 文件分别对应两种自旋的电子各自的电偶极矩;以及电场文件 efield_x.dat,x 的数量视所加的电场数量而定,这个算例就是两个:efield_0.dat 和 efield_1.dat。不考虑单位,通过下列公式得到分子的吸收谱信息:
为方向的电偶极矩,为方向的电场。程序自带了相应的后处理功能,具体请参考 tools``/``plot-tools 下的说明进行使用,其结果如下:
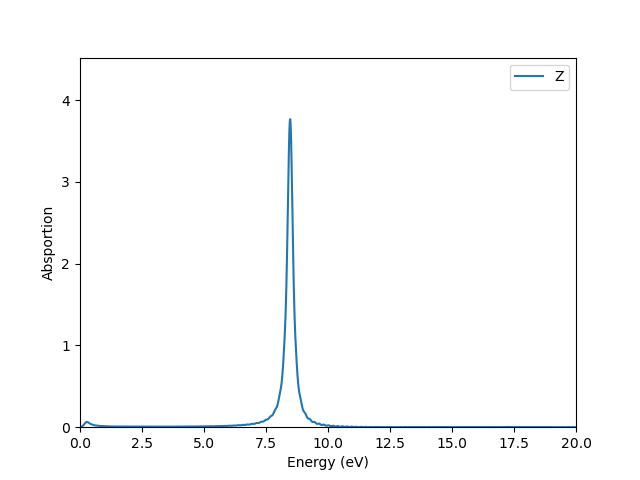
这表明在方向加的电场激发了一个频率为 8.5 eV 左右的吸收峰。
3.3 速度规范示例
算例:https://gitee.com/mcresearch/abacus-user-guide/tree/master/examples/tddft/absorption_H2_velocity
速度规范的流程与长度规范基本相同,主要不同的参数有两个:
td_stype:变为了1,代表进行速度规范计算。out_current:输出电流的开关。对无限大的周期性体系无法定义电偶极,采用电流来代替,公式可以参考本文的参考文献 2。简单来说:
因此在傅里叶变换时:
因此二者可以得到类似的结果,不过 current 的结果由于分母的原因,在极低频时会无法避免的发散。相应的问题在文献 3 中有讨论。
四、参考文献
- Meng S, Kaxiras E. Real-Time, Local Basis-Set Implementation of Time-Dependent Density Functional Theory for Excited State Dynamics Simulations[J]. The Journal of Chemical Physics, 2008, 129(5), https://doi.org/10.1063/1.2960628 .
- Pemmaraju C D, Vila F D, Kas J J, et al. Velocity-Gauge Real-Time TDDFT Within a Numerical Atomic Orbital Basis Set[J]. Computer Physics Communications, 2018, 226: 30-38, https://doi.org/10.1016/j.cpc.2018.01.013 .
- Yabana K, Sugiyama T, Shinohara Y, et al. Time-Dependent Density Functional Theory for Strong Electromagnetic Fields in Crystalline Solids[J]. Physical Review B, 2012, 85(4): 045134, https://link.aps.org/doi/10.1103/PhysRevB.85.045134 .